Si las propiedades de los materiales se pueden predecir de manera confiable, el proceso de desarrollo de nuevos productos para una amplia gama de industrias se puede simplificar y acelerar. En un estudio publicado en sistemas inteligentes avanzadosEn este estudio, investigadores del Instituto de Ciencias Industriales de la Universidad de Tokio utilizaron espectroscopia de pérdida nucleolar para caracterizar moléculas orgánicas mediante el aprendizaje automático.
Las técnicas de espectroscopia de pérdida de energía para la estructura del borde cercano (ELNES) y la estructura de rayos X del borde cercano (XANES) se utilizan para determinar información sobre los electrones y, a través de ellos, los átomos en los materiales. Tienen una alta sensibilidad y precisión y se han utilizado para seleccionar una variedad de materiales, desde dispositivos electrónicos hasta sistemas de administración de fármacos.
Sin embargo, la correlación de los datos espectroscópicos con las propiedades de un material, como las propiedades ópticas, la conductividad electrónica, la densidad y la estabilidad, sigue siendo ambigua. Se han utilizado enfoques de aprendizaje automático (ML) para extraer información de grandes conjuntos de datos complejos. Estos métodos utilizan redes neuronales artificiales, que dependen del funcionamiento de nuestro cerebro, para aprender continuamente a resolver problemas. Aunque el grupo había utilizado previamente los espectros ELNES / XANES y ML para obtener información sobre los materiales, lo que encontraron no estaba relacionado con las propiedades del material en sí. Por lo tanto, la información no se puede traducir fácilmente en desarrollos.
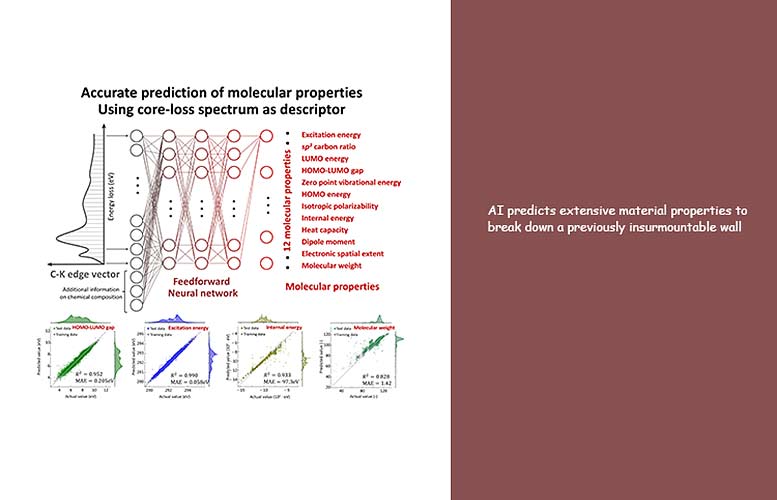
Investigadores del Instituto de Ciencias Industriales de la Universidad de Tokio están utilizando un enfoque de aprendizaje automático para predecir con éxito las propiedades de materiales no identificados previamente. Crédito: Instituto de Ciencias Industriales, Universidad de Tokio
Ahora, el equipo ha utilizado ML para revelar información oculta en espectros ELNES / XANES simulados de 22.155 moléculas orgánicas. «Los espectros ELNES / XANES de las moléculas, o sus ‘descriptores’ en este escenario, se introdujeron en el sistema», explica el autor principal, Kakeru Kikumasa. «Este descriptor es algo que se puede medir directamente en experimentos y, por lo tanto, se puede determinar con alta sensibilidad y precisión. Este método es muy útil para desarrollar materiales porque tiene el potencial de revelar dónde, cuándo y cómo aparecen ciertas propiedades de los materiales».
El modelo generado solo a partir de los espectros pudo predecir con éxito lo que se conoce como propiedades intensas. Sin embargo, no pudo predecir las propiedades de banda ancha, que dependen del tamaño molecular. Por lo tanto, para mejorar la predicción, se creó el nuevo modelo incluyendo las proporciones de tres elementos con respecto al carbono (que está presente en todas las moléculas orgánicas) como parámetros adicionales para permitir predecir correctamente amplias propiedades como el peso molecular.
«Nuestro tratamiento de aprendizaje automático de los espectros de pérdida de núcleos proporciona una predicción precisa de las propiedades de los materiales a gran escala, como la energía interna y el peso molecular. La relación entre los espectros de pérdida de núcleos y las propiedades generales no existía antes; sin embargo, la inteligencia artificial pudo revelar lo oculto “Nuestro enfoque también se puede aplicar para predecir nuevas propiedades y funciones de materiales”, dice el autor principal Teruyasu Mizoguchi. «Creemos que nuestro modelo será una herramienta muy útil para el desarrollo de materiales de alto rendimiento en una amplia gama de industrias».
Referencia: «Cuantificación de las propiedades de moléculas orgánicas utilizando espectros de pérdida fundamental como descripción de una red neuronal», 15 de octubre de 2021, sistemas inteligentes avanzados.
DOI: 10.1002 / aisy.20210103

«Erudito en viajes incurable. Pensador. Nerd zombi certificado. Pionero de la televisión extrema. Explorador general. Webaholic».