
Google DeepMind
La mayoría de las actividades que tienen lugar dentro de las células (las actividades que nos hacen vivir, respirar y pensar como animales) están a cargo de las proteínas. Permiten que las células se comuniquen entre sí, impulsan el metabolismo básico de la célula y ayudan a convertir la información almacenada en el ADN en más proteínas. Todo depende de la capacidad de la cadena de aminoácidos de la proteína para plegarse en una forma tridimensional compleja pero específica que le permita realizar su función.
Hasta esta década, comprender la forma tridimensional significaba purificar una proteína y someterla a un proceso que requería mucho tiempo y esfuerzo para determinar su estructura. Pero eso cambió con el trabajo de DeepMind, una división de inteligencia artificial de Google, que lanzó Alpha Fold en 2021, y un esfuerzo académico similar poco después. El programa no fue perfecto. Tuvo problemas con proteínas más grandes y no proporcionó soluciones de alta confianza para cada proteína. Pero muchas de sus predicciones resultaron ser sorprendentemente precisas.
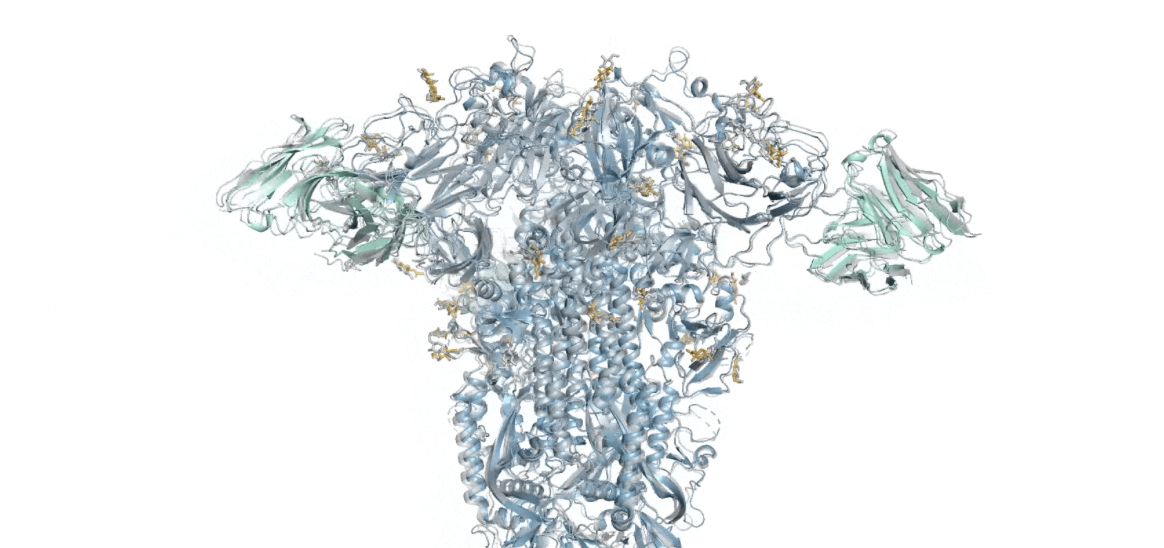
Sin embargo, estas estructuras sólo cuentan la mitad de la historia. Para realizar su trabajo, casi todas las proteínas deben interactuar con algo más, como otras proteínas, ADN, sustancias químicas, membranas y más. Si bien la versión inicial de AlphaFold pudo manejar algunas interacciones proteína-proteína, el resto siguieron siendo cajas negras. DeepMind anuncia hoy la disponibilidad de AlphaFold Versión 3, en la que partes de su motor central se han modificado significativamente o se han reemplazado por completo. Gracias a estos cambios, el software ahora maneja muchas interacciones y modificaciones de proteínas adicionales.
cambiar piezas
El AlphaFold original se basaba en dos funciones de software principales. Uno de ellos tuvo en cuenta los límites evolutivos de la proteína. Al observar la misma proteína en múltiples especies, se pueden identificar partes que son siempre iguales y, por lo tanto, probablemente centrales para su función. Esta centralidad significa que es probable que siempre estén en la misma ubicación y orientación en la estructura proteica. Para hacer esto, el AlphaFold original encontró tantas versiones de la proteína como fuera posible y las secuenció para buscar partes que mostraran poca diferencia.
Sin embargo, hacerlo es costoso desde el punto de vista computacional, ya que cuantas más proteínas alinees, más restricciones tendrás que resolver. En la nueva versión, el equipo de AlphaFold todavía identifica varias proteínas relacionadas, pero ha pasado a realizar alineaciones utilizando en gran medida pares de secuencias de proteínas dentro del grupo de proteínas relacionadas. Puede que esto no sea tan informativo como los alineamientos múltiples, pero es más eficiente desde el punto de vista computacional y la información que falta no parece ser tan importante para descubrir estructuras de proteínas.
Utilizando esta alineación, un módulo de software independiente pudo detectar relaciones espaciales entre pares de aminoácidos dentro de la proteína objetivo. Luego, estas relaciones se traducen en coordenadas espaciales para cada átomo mediante un código que tiene en cuenta algunas de las propiedades físicas de los aminoácidos, como qué partes del aminoácido pueden rotarse en relación con otras partes, etc.
En AlphaFold 3, la predicción de las posiciones atómicas se maneja mediante un módulo de difusión, que se entrena dándole una estructura conocida y versiones de esa estructura donde se ha agregado ruido (en forma de posiciones cambiantes de algunos átomos). Esto permite que el módulo de difusión tome ubicaciones imprecisas descritas por posiciones relativas y las convierta en predicciones precisas de la ubicación de cada átomo en la proteína. No necesita conocer las propiedades físicas de los aminoácidos, porque puede descubrir qué hacen normalmente observando suficientes estructuras.
(DeepMind tuvo que entrenar en dos niveles diferentes de ruido para ejecutar el módulo de difusión: uno en el que se cambiaban las posiciones de los átomos dejando intacta la estructura general y un segundo en el que el ruido implicaba cambiar la estructura a gran escala del átomo. . La proteína, que afecta la ubicación de muchos átomos.)
Durante el entrenamiento, el equipo descubrió que se necesitaban alrededor de 20.000 copias de estructuras de proteínas para que AlphaFold 3 consiguiera que aproximadamente el 97 por ciento del conjunto de estructuras de prueba fuera correcto. 60.000 veces, comenzó a corregir las interfaces proteína-proteína también a esa frecuencia. Es importante destacar que también comenzó a ensamblar correctamente proteínas con otras moléculas.

«Organizador. Escritor. Nerd malvado del café. Evangelista general de la comida. Fanático de la cerveza de toda la vida.






{kind=link}